Two-Photon Microscopy
Introduction
Two-photon microscopy is a technique that avoids the limitations of traditional fluorescence microscopy. Typical fluorescence microscopy involves using illumination of a specific wavelength in order to excite fluorophores within a sample. However, standard widefield epifluorescence imaging also collects fluorescence from outside the focal plane, resulting in background illumination and image degradation.
Confocal microscopes use a pinhole to block all emission fluorescence except the focal plane, eliminating out of focus light and improving the image quality. This technique also allows for optical sectioning, imaging 2D planes within the sample. However, this breaks down when looking at particularly thick samples, as the light can only penetrate a short distance into the sample and microscope objectives are physically limited by their working distance. The deeper the desired viewing plane, the greater the proportion of light that is absorbed/scattered.
A solution to issues found in both widefield and confocal microscopy is found in two-photon microscopy, which images using excitation in the near-infrared portion of the spectrum.
Imaging In The Near-Infrared
The impact of absorption and scattering can be avoided by imaging at different wavelengths. While typical epifluorescence and confocal microscopy are done with visible light (~400-700 nm), there are several advantages to using larger wavelengths of light for imaging, such as near-infrared (NIR), which occupies ~700‑1700 nm, as seen in Fig.1.
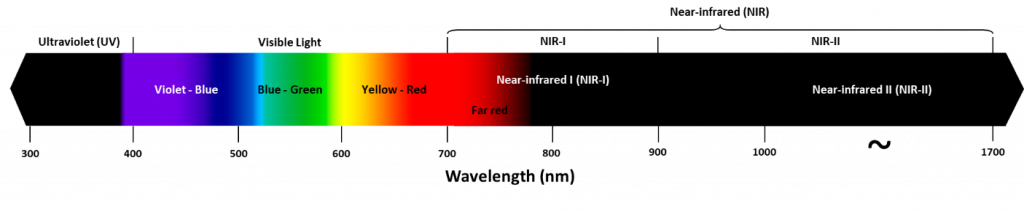
Figure 1: The electromagnetic spectrum, ranging from UV to NIR. NIR occupies the 700-1700 nm range, consisting of far red (700-780 nm), NIR-I (700-900 nm) and NIR-II (900-1700 nm). The greater the wavelength, the lower the frequency and energy of the photons. This means that NIR photons have a lower energy than visible light photons.
Both absorption and scattering of light change with wavelength, as seen in Fig.2, which demonstrates the absorption coefficients for substances commonly found in tissues (water, lipids, DNA). The absorption coefficient essentially refers to how far into a material light can penetrate before it is absorbed. By penetrating further into a sample, light can be used to image through thicker samples with optical sectioning.
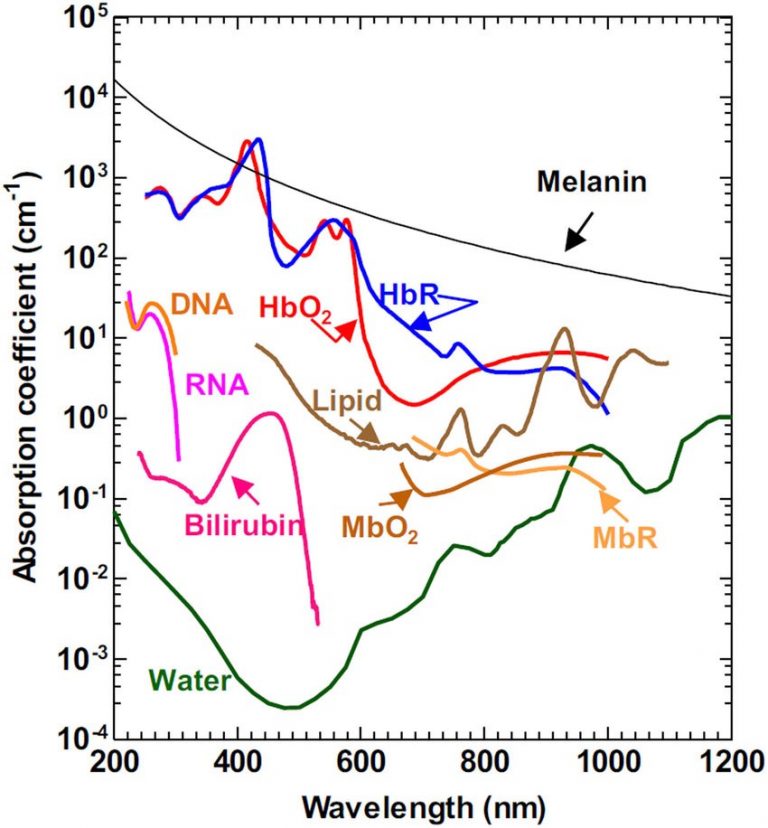
Figure 2: Absorption coefficients relative to wavelength for common substances found in biological tissues. The absorption coefficient refers to how far into a material light can penetrate before it is absorbed. Bilirubin: yellow compound produced by breaking down heme-iron from red blood cells, HbR: hemoglobin, HbRO2: Oxyhemoglobin, MbR: Myoglobin, MbRO2: Oxymyoglobin. Image from Yao and Wang (2013).
The graph in Fig.2 demonstrates that there is a greater absorption coefficient of these biological substances in the NIR, particularly water. While water is transparent to visible light, it absorbs in the NIR (~700-1700 nm), this means that when water is imaged in the NIR it appears as black, this can be seen in Fig.3. This phenomenon also explains why, if you shine a bright light up to your hand only red light will come through, no green or blue is seen, due to the absorbance of tissues dropping off towards the red and NIR, also seen in Fig.3.

Figure 3: Imaging in the red/NIR part of the spectrum. The top two images show a glass of water, a visible light image on the left and a NIR image on the right. As water absorbs increasingly more beyond wavelengths of 700 nm, it appears as a dark liquid in the NIR. The bottom image shows a white light shone through a hand, only the red light passes through, revealing some structures within. Bones and other details are not seen due to light scattering through this arbitrarily thick sample. Images from Wikimedia Commons.
This makes NIR an effective series of wavelengths with which to image biological tissues. Using NIR photons decreases absorption from thick samples, allowing the light to penetrate deeper into these samples. Using NIR also reduces scattering of light, as Rayleigh scattering is wavelength dependent, the equation being 1/λ4. Rayleigh scattering follows an inverse relation of λ-ω where λ is wavelength and ω is the wave exponent parameter. The longer the wavelength, the less the light scattering occurs. Overall, imaging in the NIR results in better quality images when using thicker samples such as whole organisms.
In order to image in the NIR, certain infrared dyes can be used (excited at 700-800 nm and fluoresce around 900-1000 nm, have had success in whole animal imaging), but another approach is to image standard fluorescent dyes in the NIR by using two-photon microscopy. This two-photon approach allows a standard green fluorescent dye to be excited using two NIR photons, allowing for a much wider and more familiar range of fluorescent dyes to be used, while taking advantage of the lower scattering and absorption of NIR imaging.
Two-Photon Excitation
Conventional excitation in fluorescence microscopy typically involves one photon. Fluorescent molecules (fluorophores) typically exist in the ground state (lower energy, S0) and become excited when exposed to photons of a certain wavelength, moving to a higher energy state (Sn). After moving to a higher energy state, there is some internal conversion and relaxing where the molecules can drop in energy slightly (to S1), but eventually, these fluorophores will drop back to the ground state by releasing the energy as emitted photons, this is the mechanism behind fluorescence.
Due to the move from Sn to S1, the emitted photon is of lower energy than the excitation photon and therefore has a longer wavelength, thus appearing as a different color. When excited with blue light, a fluorophore can emit green light (green photons have less energy and longer wavelength, see Fig.1).
While NIR imaging is advantageous, NIR photons have a much longer wavelength than visible light photons (700-900 nm) and consequently have much lower energy. This means a single NIR photon does not have sufficient energy in order to bump a fluorophore from S0 to Sn, and no fluorescence will occur. But if one NIR photon isn't enough for excitation, what about two?
Fig.4 shows the difference between single-photon excitation and two-photon excitation. While a single 400 nm blue photon is enough to excite a fluorophore, it takes two 800 nm NIR photons delivered in quick succession in order to excite the same fluorophore.
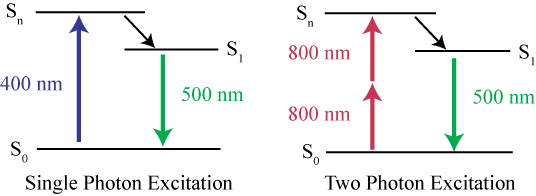
Figure 4: Jablonski energy diagrams for single-photon and two-photon excitation. On the left is single-photon excitation, where a single 400 nm photon is sufficient to cause fluorescence. On the right is two-photon excitation, where two 800 nm NIR photons are needed to cause fluorescence. Image from Nowak et al. (2010).
The two NIR photons must arrive in extremely quick succession for this to work, both photons must arrive nearly simultaneously, within less than one femtosecond (0.000001 nanoseconds or 10-15 seconds) of each other. By arriving this close together in time, the energy of the two photons is summed and is sufficient to excite fluorophores. This is two-photon (or multi-photon) excitation, where NIR photons can be used with standard fluorescent dyes, delivering the best of both worlds.
Localized Excitation
In single-photon excitation, the relationship between excitation light intensity and fluorescence intensity is linear, as a single photon can cause fluorescence. In two-photon excitation, the relationship is non-linear (quadratic) as the fluorescence intensity is dependent on the square of the number of photons arriving at the fluorophore due to there being multiple photons involved (the probability of absorbing the first photon multiplied by the probability of absorbing the second). This nonlinearity of excitation is quite advantageous for two-photon, as it causes localized excitation, seen in Fig.5.

Figure 5: Simultaneous single (1P, linear) and two-photon (2P, non-linear)excitation within a cuvette. The left objective is illuminating thecuvette with single-photon excitation, the light is absorbed throughoutthe entire light path. The right objective is illuminating the cuvettewith two-photon excitation, the only location where fluorescence isseen is the small dot in the center of the sample (highlighted with white arrow). Photographed by Amos W.B. and Cipollone M, MRC Cambridge.
As there is only a small area where the laser beam is focused enough to deliver two photons at near‑simultaneous speeds, two-photon excitation is highly localized. This means that there is no out‑of-focus light as the sample is only excited at the focal plane, similarly to confocal microscopy. Not only does two-photon excitation allow for use of the suite of standard fluorescent dyes with NIR excitation (resulting in less absorption and scattering), it also achieves the same point-spread function (PSF) as a confocal microscope without the need for a pinhole. The small point of excitation is scanned across large samples, similarly to laser scanning confocal microscopy, resulting in an image.
Due to the fact that the fluorescence intensity is dependent on the square of the excitation intensity, two‑photon excitation benefits from high excitation power. Pulsed lasers are used to get high peak power but normal average power, making them suitable for two-photon microscopy.
Applications Of Two-Photon Microscopy
One of the main advantages of two-photon microscopy is the ability to image very thick samples. While standard single-photon confocal can only image samples up to 200 μm thickness, two-photon microscopy can image samples up to several millimeters thickness and is less limited in sample choice.
Two-photon microscopy is also useful when imaging live samples as it can image cellular detail in live animals without the need for fixative, clearing, or slices of sections, optical sectioning can be performed within live samples. Typical fluorescence microscopy has to combat decreasing fluorescence levels with increasing image depth (due to scattering from the sample) but with two-photon, fluorescence remains stable even with increasing image depth. This is useful for many imaging applications, including such fields as immunology and neuroscience, as cells can be observed moving around deep within live animals.
An example of this ability to image thick samples from live specimens is displayed in Fig.6, showing images from a mouse brain up to several millimeters in depth.
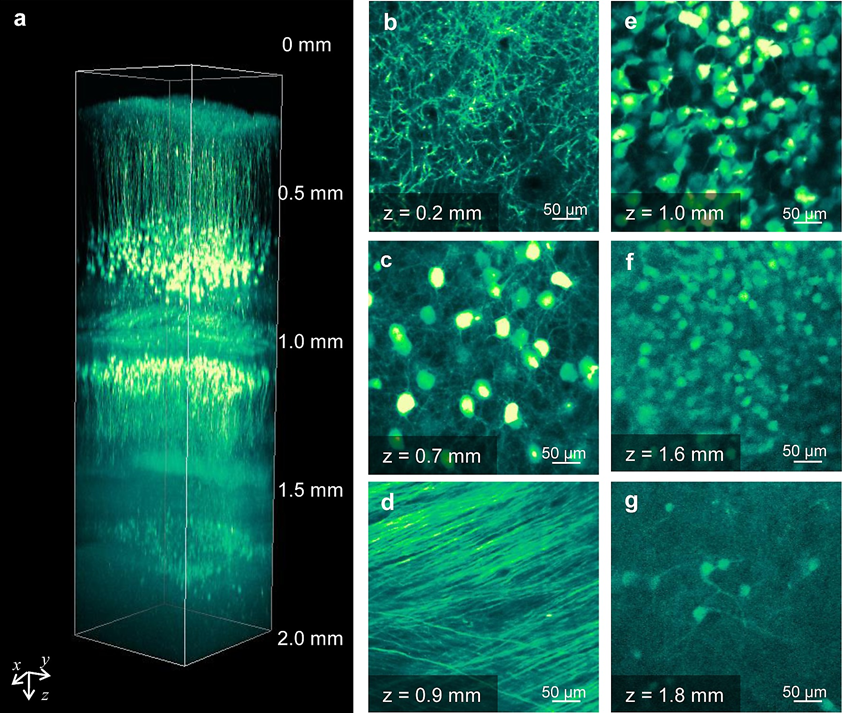
Figure 6: Two-photon deep imaging of a whole mouse brain. A) 3D XYZ reconstruction of a whole mouse brain, this section is 2 mm in depth. B-G) XY images at different depths from different regions of the mouse brain, including B) layer V neuron apical dendrites, C) layer V neuron cell bodies, D) white matter, E) hippocampal CA1 cell bodies, F) hippocampal dentate gyrus upper blade, and G) lower blade. Image from Aoyagi et al. (2015).
Two-photon also has applications when using UV-excitable fluorophores, as the NIR excitation is less harmful to samples with UV-excitable fluorophores when compared to using UV light.
Cameras For Two-Photon Microscopy
Camera requirements for two-photon microscopy are similar to other microscopy applications such as confocal. Cameras should feature high sensitivity, with a high quantum efficiency (QE) across a wide range of photon wavelengths, in order to detect signals from deep within live samples. As larger samples can be used with two-photon, it should be paired with cameras with a large field of view (FOV) in order to efficiently capture emission from large portions of the sample. High speeds and a high resolution are also desirable, in order for the detector to be useful with the wide range of samples a two-photon imaging system is likely to be used with.
Summary
Two-photon excitation microscopy is an elegant fluorescent imaging technique that allows imaging of large samples at depths of several millimeters, featuring less phototoxicity and being suitable for imaging of live samples. Two-photon imaging involves the use of two photons in the near-infrared arriving almost simultaneously, in order to have the same energy as a single photon of a lower wavelength. Due to a small excitation area, two-photon microscopy features many of the same advantages as confocal systems, including elimination of out-of-focus light and optical sectioning of samples.
References
1. Yao J. and Wang L.V. (2013) Photoacoustic microscopy, Laser Photon Review, 7(5) pp.758-778.
2. Nowak D.B., Lawrence A.J., Sanchez E.J. (2010) Apertureless near-field/far-field CW two-photon microscope for biological and material imaging and spectroscopic applications, Applied Optics 49(35).
3. Aoyagi Y, Kawakami R, Osanai H, Hibi T, Nemoto T (2015) A Rapid Optical Clearing Protocol Using 2,2′-Thiodiethanol for Microscopic Observation of Fixed Mouse Brain. PLoS ONE 10(1): e0116280. https://doi.org/10.1371/journal.pone.0116280
Want To Learn More?
Back To Imaging Topics
Book A Camera Course
Knowledge and Learning Hub