Multiomics
Introduction
The phenotype of an organism is determined by the central dogma of molecular biology that states, "DNA makes RNA, and RNA makes protein" (Crick, 1970). These "omes" have been studied for a number of years in the fields of genomics (DNA), transcriptomics (RNA) and proteomics (proteins) as researchers look to understand the flow of information from genetic causes of disease to the functional consequences (Figure 1).
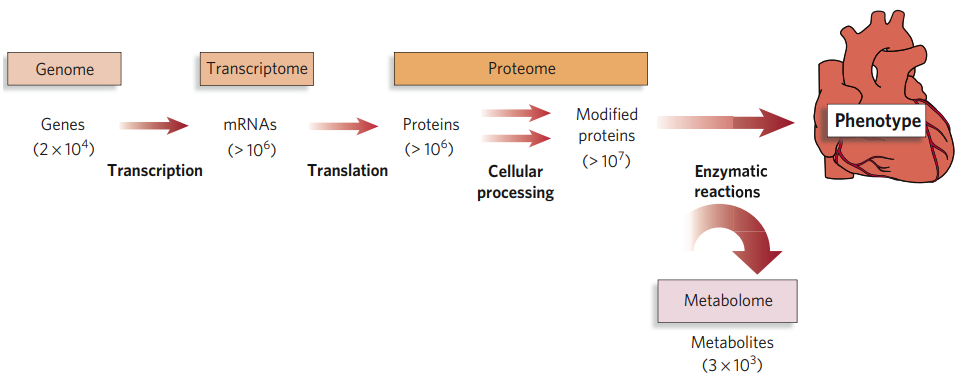
Figure 1: The conceptual relationship of the genome, transcriptome, proteome and metabolome (Gerszten, 2008)
Adding "omics" to a molecular term simply implies that all molecules of that particular type are studied comprehensively. In the case of genomics for example, an organism's whole genome is studied. This differs from genetics which is the study of individual genes.
Advances in "omics" technology over the years has resulted in instrumentation and techniques used to study them becoming more and more affordable in both time and cost. Furthermore, the ability to perform them in a high throughput manner means that researchers have access to more data than ever before.
This has led inevitably to an approach that draws together data from multiple "omic" techniques - multiomics. The growth in multiomic approaches has been tremendous in recent years as it's becoming clear that one set of omics data is not enough to make causative conclusions, an issue that integrating multiple omics types is helping to overcome.
Genomics
Studying the genome is essential for human health and medicine because an individual's genetic background is an important determinant of susceptibility to certain diseases and conditions. Methods used to study the genome are typically either sequencing studies or spatial studies (or a combination of both).
Sequencing studies focus on identifying and characterizing DNA sequences known to play a role in disease whereas spatial studies use imaging techniques such as fluorescence microscopy to spatially locate specific DNA sequences on chromosomes in a variety of cell types.
DNA Sequencing
There are currently three main sequencing methods used to perform genomic analysis:
- Sanger sequencing
- DNA-microarrays
- Next-generation sequencing (NGS)
The Sanger method (Sanger et al. 1977) is a base-by-base sequencing method that was most famously used to deduce the human genome in the Human Genome Project. As the Human Genome Project progressed, improvements were made, and automated sequencing machines were manufactured to perform the Sanger method at reduced cost and improved speed which resulted in the early completion of the Human Genome Project in April 2003.
Since then, easier and faster methods have been developed using different methods such as the hybridization strategies used by DNA-microarrays and NGS methods.
DNA-microarrays are used to measure the expression of large numbers of genes or to determine the sequence of bases of multiple genome regions. A DNA-microarray (also known as a DNA chip or biochip) is a solid surface onto which microscopic DNA spots are attached. These DNA spots comprise known DNA sequences typically called "oligos" (short for oligonucleotide). The sample DNA being investigated is introduced to the microarray where it binds to complementary oligo sequences. The strength of the fluorescence signal (intensity) from the sample DNA bound to the spot can be compared with the same spot under different conditions to infer meaning from relative expression levels (Figure 2).
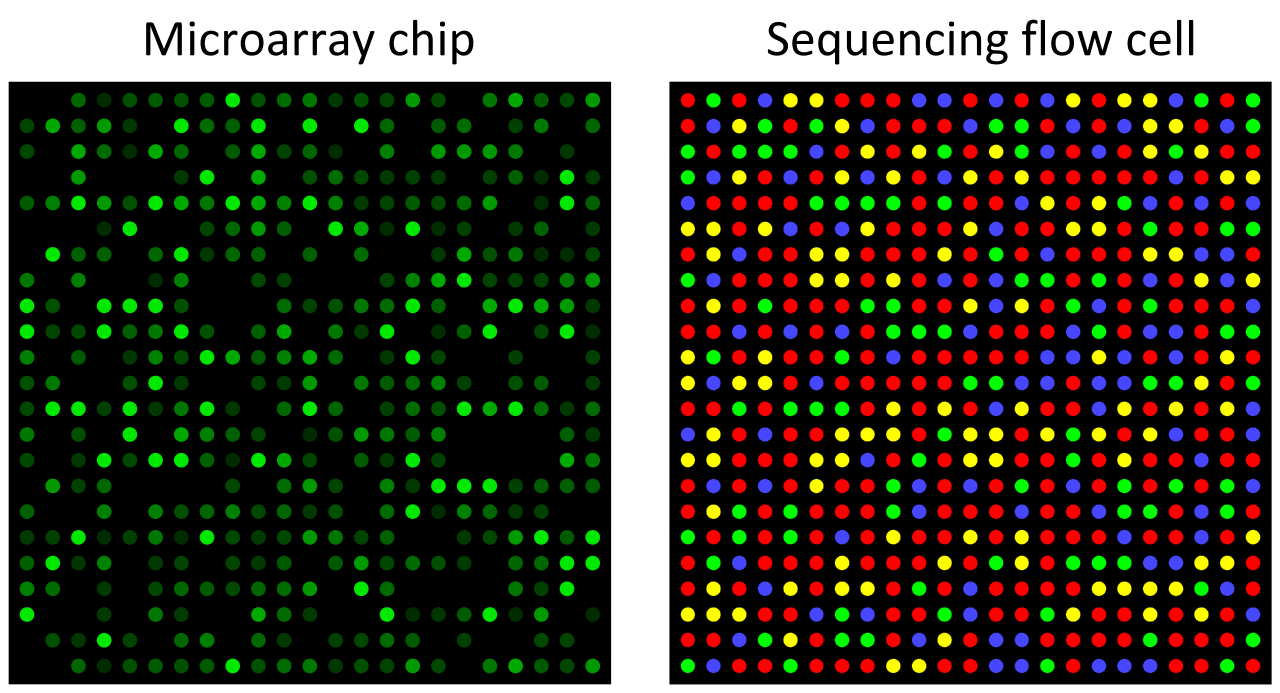
Figure 2: Microarray and sequencing flow cell. In a microarray chip, each spot on a chip is a defined oligonucleotide probe, and fluorescence intensity directly detects the abundance of a specific sequence. In a high-throughput sequencing flow cell, spots are sequenced one nucleotide at a time, with the colour at each round indicating the next nucleotide in the sequence. Image by Thomas Shafee, distributed under a Creative Commons Attribution 4.0 International license.
One of the downsides of DNA-microarrays is that the DNA sequence of interest must be known in advance. NGS strategies overcome this issue and can be used for sequencing of unknown DNA fragments.
There are many different NGS strategies, a discussion of which is outside the scope of this article, but they all broadly use the same technique of fragmenting DNA into pieces which are then all sequenced and aligned to a reference sequence. In this way, millions of DNA fragments are sequenced in parallel and are often sequenced multiple times for accuracy.
This technique therefore allows large amounts of unknown DNA to be sequenced in a very short amount of time with highly accurate results.
Spatial DNA Studies
It's possible to localize DNA within the genome using a technique called Fluorescence In Situ Hybridization (FISH).
In this approach, a fluorescent section of DNA is created which is complementary to the DNA sequence of interest in the cell sample. The fluorescent DNA is incubated with the full set of chromosomes from the cell sample on a microscope slide. The fluorescently labeled DNA finds its matching segment on one of the chromosomes, where it binds.
Using fluorescence microscopy, the DNA sequence of interest can then be located in the cell which allows the researcher to identify the location of this section of DNA (Figure 3).
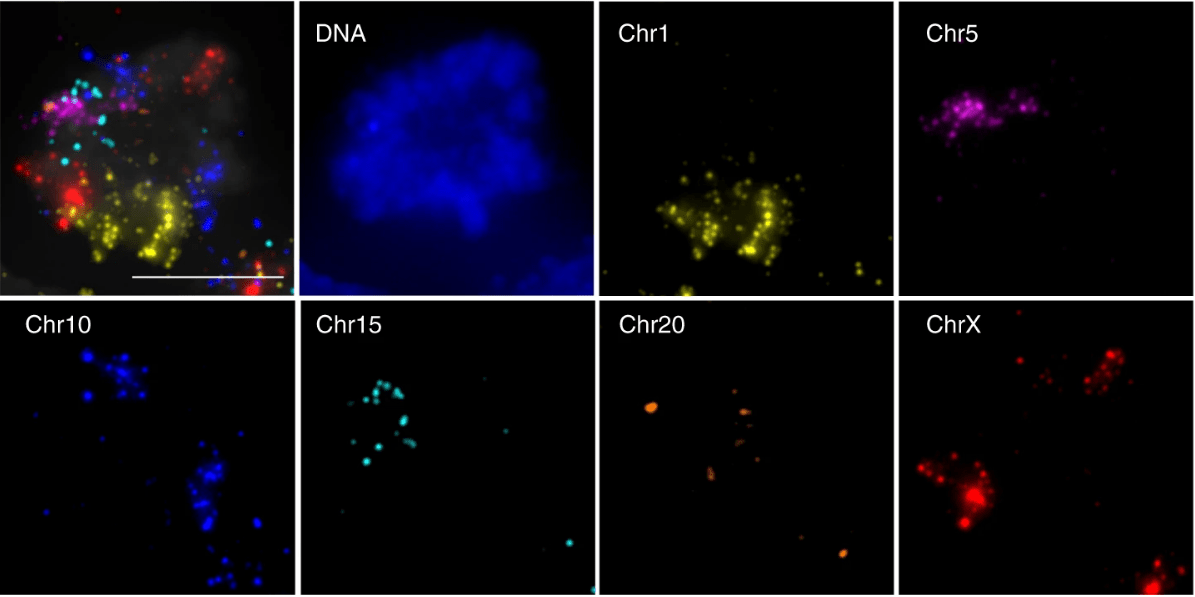
Figure 3: Maximum intensity Z-projections of the location of six different chromosomes in mototic hESCs using chromosome-spotting probes. Gelali et al. 2019
Transcriptomics
The transcriptome comprises all RNA transcripts in the cell including all coding RNAs (mRNA) and non-coding RNAs. Typically, researchers are only interested in mRNA analysis because an mRNA molecule corresponds to the genetic sequence of a specific gene which it carries to the ribosome for protein synthesis. As such, it provides the most direct insight into gene expression.
Like genomics, transcriptomics uses both sequencing and spatial studies.
RNA Sequencing
Similar to the previous section on DNA sequencing, RNA is sequenced using both microarray and next generation sequencing techniques. As with DNA, microarrays are generally cheaper but require prior knowledge of the sequence of interest whereas NGS techniques allow for discovery of unknown sequence information.
Information gained from RNA sequencing techniques provides a unique snapshot of the transcriptomic status of a disease and investigating an unbiased population of transcripts allows for the identification of novel transcripts, fusion transcripts and non-coding RNAs that could be undetected with different technologies. This has significant potential in medicine with the ability to identify new disease biology, profile biomarkers for clinical indications, infer druggable pathways, and make genetic diagnoses.
Spatial RNA Studies
RNA-FISH is an adaptation of FISH that is used in a similar way to fluorescently label specific RNA sequences, most often used to identify specific genes. Visualizing specific gene locations in cells provides a wealth of information about cell or tissue specific gene expression.
Figure 4 shows one such example of FISH being used to differentiate cell types in the retina based on the difference in gene expression of these cell types.
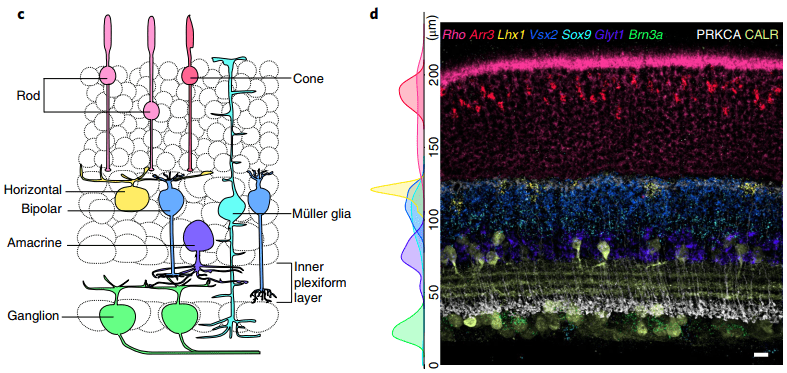
Figure 4: Right FISH detection of specific retinal cell types Left Schematic of retinal cell classes detected. Kishi et al. 2019
One significant challenge with visualizing multiple genes in the same image is a lack of spectral separation needed to identify more than 4 genes at once. In fluorescence microscopy it's fairly common to use the four colors; blue, yellow, red, and far red. However, spatial transcriptomics is used to visualize hundreds or even thousands of genes which cannot be spectrally separated in a single image.
Because of this, spatial transcriptomics makes use of wash cycles to image a certain number of fluorophores per cycle which are then washed out and new fluorophores introduced until all genes have been visualized. The image in Figure 4 for example was put together from three FISH detection cycles to identify seven retinal cell populations. For this reason, instruments for the automated preparation and visualization of FISH cycles are commonly used.
Proteomics
The proteome is the entire set of proteins present in a cell or tissue. Studying the proteome can therefore be used to investigate:
- When and where proteins are expressed
- Rates of protein production, degradation, and steady-state abundance
- The movement of proteins between subcellular compartments
- The involvement of proteins in metabolic pathways
- How proteins interact with one another
- How proteins are modified (for example, post-translational modifications (PTMs) such as phosphorylation)
However, proteomics comes with a considerable degree more complexity than genomics or transcriptomics as the 4-base code used in DNA and RNA is translated into a code of 20 amino acids which make up all proteins. In addition, proteins can be folded in a large number of different ways, chemically modified (such as through phosphorylation and glycosylation) and can have multiple isoforms.
Due to the complexity, investigating protein sequences can't be performed as well at the omics level like with DNA and RNA. Some additional reasons for this are:
- Reference databases of protein sequences are incomplete or inaccurate
- Mass spectrometry (used to identify protein sequences) is biased towards higher concentration peptides
- Protein sequences can't easily infer protein function as this depends so much on how the protein is folded
- Differences in methodology across research groups means there is no uniform protocol for preparation or analysis
Nevertheless, proteomics has been used successfully to investigate many biological problems such as identifying which proteins interact with key disease proteins, such as tumor suppressor protein p53, or localizing proteins to specific cellular compartments like the mitochondria.
Protein Antibody Detection
Proteins have long been detected using antibodies in techniques such as immunoassays, protein purification and fluorescence microscopy and is one of the most common molecular biology tools.
In particular, the enzyme-linked immunosorbent assay (ELISA) has been used for decades to detect and quantitatively measure proteins in samples and the western blot has long been used for detection and quantification of individual proteins. Tagging proteins with primary and secondary antibodies is also a well-used technique for protein imaging which allows proteins to be visualized in cells and tissues and tracked in three dimensions using advanced microscopy techniques to provide spatial information.
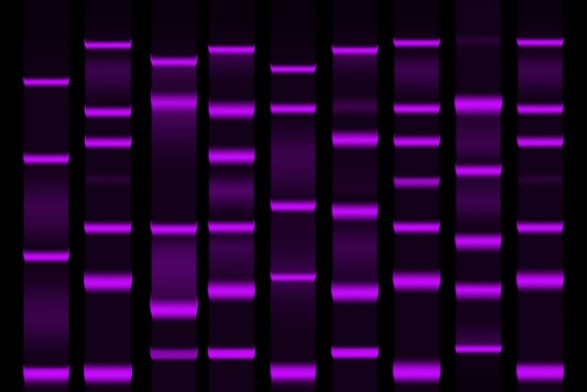
Figure 5: Gel electrophoresis separation electrophoretogram
Multiomics
The fields of genomics, transcriptomics and proteomics are not isolated biological systems, they are all fundamentally associated with each other in a complex system that represents the true biological environment.
In an age of ever-increasing data collection and analysis capability, one set of omics data alone is no longer enough to make causative conclusions. Integration of different omics data types is now often being used to build up the full picture - this is multiomics.
As an example, in the past, mRNA analysis was used to infer which proteins were produced by the cell but this was found to not actually correlate with protein content. It is now known that mRNA is not always translated into protein. The amount of protein produced for a given amount of mRNA depends on the gene it is transcribed from but also the physiological state of the cell. Considering this disparity, and that proteins are the molecules that control almost all cellular processes, the benefit from integrating these technologies for a more complete image is clear.
However, despite the advantages of multiomics there are some significant challenges. Firstly, the integration of data sets generated in multiomics research is by no means trivial. Each omics technology naturally consists of different types of data which complicates the analysis to begin with. This is further complicated by the sheer volume of data produced by these high throughput techniques. Transforming the different data sets and combining them is therefore an ongoing problem to be solved.
The second problem is one of space. Spatial multiomics studies may plan to image hundreds or thousands of genes and proteins together. The result is that a large number of fluorophores end up occupying the same area in the cell which makes resolving individual genes or proteins in this area difficult.
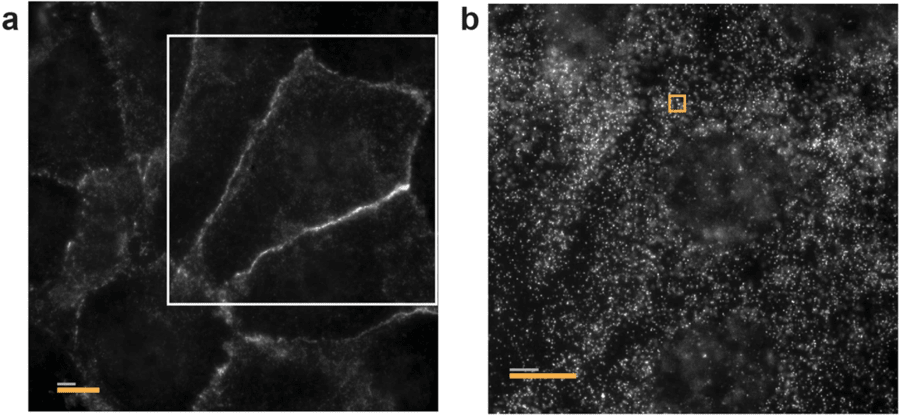
Figure 6: (a) Image of an expanded U-2 OS sample stained with MERFISH encoding probes for 129 RNA species, cadherin primary antibodies, oligo-conjugated secondary antibodies and visualized with a Cy5-conjugated complementary probe that detects the secondary antibodies. (b) Image of the same expanded sample visualized with a Cy5-labeled readout probe that detects one of the bits of the RNA barcodes at one focal plane for the white boxed region in (a). Wang et al. (2018)
The obvious solution would be to separate all fluorophores temporally, but this means greatly increasing the number of cycles which greatly increases the time taken to image them all. Moving from four fluorophores per image to one fluorophore per image for example would increase the number of cycles 4-fold which is substantial when hundreds or thousands of molecules need to be imaged.
Another solution would be to use super-resolution techniques such as expansion microscopy (Figure 6) and localization microscopy which physically separates the molecule further apart. These methods have been used very successful but add complexity to the sample preparation and not all samples are compatible with these methods.
A lot of ground has been covered in recent years but solving these problems is essential to the success of future multiomic approaches.
Summary
Multiomics allows researchers to obtain a greater depth of knowledge than what is possible through a single omics study. By combining genomic, transcriptomic and proteomic datasets, a greater understanding of complex populations and processes can be obtained which should drive improved clinical diagnostics and therapies.
Multiomics is still a young field but the number of publications citing it is increasing massively as the potential is realized. Along with this, instrumentation and technology are developing rapidly and increasing in capability which should allow for even greater progress in this field in the coming years.
References
Beale, D. J., Karpe, A. V. & Ahmed, W. (2016) Beyond Metabolomics: A Review of Multi-Omics-Based Approaches. In: Beale, D., Kouremenos, K., Palombo, E. (eds) Microbial Metabolomics. Springer, Cham. https://doi.org/10.1007/978-3-319-46326-1_10
Crick, F. (1970) Central Dogma of Molecular Biology. Nature. Vol 227, August 8.
Gelali, E., Girelli, G., Matsumoto, M., Wernersson, E., Custodio, J., Mota, A., Schweitzer, M., Ferenc, K., Li, X., Mirzazadeh, R., Agostini, F., Schell, J. P., Lanner, F. Crosetto, N. & Bienko, M. (2019) iFISH is a publically available resource enabling versatile DNA FISH to study genome architecture. Nature Communications, 10(1). doi:10.1038/s41467-019-09616-w
Gerszten, R. E. & Wang, T. J. (2008) The search for new cardiovascular biomarkers. Nature. Vol 451, 12 February.
Manzoni, C., Kia, D. A., Vandrovcova, J., Hardy, J., Wood, N. W., Lewis, P. A. & Ferrari, R. (2016) Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Briefings in Bioinformatics. 19(2), 2018, 286-302
Sanger, F., Nicklen, S., Coulson, A. R. (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 1977;74:5463-7.
Wang, G., Moffitt, J. R. & Zhuang, X. (2018) Multiplexed imaging of high-density libraries of RNAs with MERFISH and expansion microscopy. Scientific Reports. Volume 8, Article number: 4847
Further Reading
Back To High Content Imaging
Join Knowledge and Learning Hub